


Effect of Gene Co-expression on Heterologous Soluble Expression,Purification and Enzymatic Properties of Human LysoPLD
-
摘要: 目的:为实现人源溶血磷脂酶D(LysoPLD)的原核异源可溶性表达。方法:通过NCBI检索,确定人源LysoPLD基因序列(GenBank: L46720.1)。采用密码子优化后的序列,克隆至pET-28a表达载体中,采用共表达麦芽糖结合蛋白融合标签(MBP)和共表达促蛋白正确折叠分子伴侣触发因子Trigger factor(tig)两种方式提高LysoPLD蛋白在大肠杆菌中的异源可溶性表达,建立对应蛋白的纯化工艺包括离子柱纯化,硫酸铵盐析,疏水柱纯化,淀粉树脂柱(Amylose Resin)纯化,分离纯化获得的重组酶,经聚丙烯酰胺凝胶电泳(SDS-PAGE)测定蛋白纯度,以对羟基棕榈酸酯为底物对比两种蛋白的酶学性质。结果:成功构建载体pET28a-MBP-LysoPLD和pET28a-pTF16-LysoPLD,并获得工程菌BL21(DE3)-pET28a-MBP-LysoPLD和BL21(DE3)-pET28a-pTf16-LysoPLD。BL21(DE3)-pET28a-MBP-LysoPLD经0.6 mmol/L异丙基硫代半乳糖苷(IPTG)低温诱导过夜可获得上清表达的MBP-LysoPLD蛋白;BL21(DE3)-pET28a-pTf16-LysoPLD在含有0.5 μg/mL L-Arabinose的LB培养基中培养,经0.1 mmol/L IPTG低温诱导表达,可获得可溶性表达的LysoPLD蛋白,经纯化,酶纯度可大于80%。以对羟基棕榈酸酯为底物,对比两种方法得到的蛋白的酶学性质,发现二者催化反应的最适温度、最适pH、最适Ca2+浓度、比酶活基本一致。结论:两种基因共表达方式都可实现人源LysoPLD的在大肠杆菌中的可溶性表达,且酶学性质基本相同。
-
关键词:
- 人源溶血磷脂酶D /
- 异源可溶性表达 /
- 麦芽糖结合蛋白融合标签 /
- 触发因子 /
- 酶学性质
Abstract: Objective:This study aimed to express soluble human LysoPLD in Escherichia coli.Methods:Human LysoPLD gene sequence (GenBank: L46720.1) was retrieved from NCBI and synthesized chemically. The codon optimized sequence was cloned into pET-28a vector and transformed to the E.coli BL2(DE3) strain which was co-expressed with maltose binding protein fusion tag (MBP) or co-expressed with a pTF16 chaperone plasmid, then induced by isopropyl thiogalactoside (IPTG). The downstream purification process was established, including ion column elution, ammonium sulfate salting out, hydrophobic column purification, and amylose resin purification. The LysoPLD purity was determined by polyacrylamide gel electrophoresis (SDS-PAGE) and the enzymatic properties were determined by catalytic reaction of p-hydroxypalmitate.Results:The plasmids pET28a-MBP-LysoPLD and pET28a-pTf16-LysoPLD were constructed, then transformed into the E.coli BL21(DE3) strain respectively. BL21(DE3)-pET28a-MBP-LysoPLD was induced by 0.6 mmol/L IPTG, and incubated overnight to obtain MBP-LysoPLD protein; BL21(DE3)-pET28a-pTF16-LysoPLD was cultured in LB medium containing 0.5 µg/mL L-arabinose then induced by 0.1 mmol/L IPTG, the soluble LysoPLD was obtained. After downstream purification, the purity of the human LysoPLDs expressed through both methods were more than 80%. It was found that using p-hydroxypalmitate as substrate, the optimum reaction conditions including optimal temperature, pH and Ca2+ concentration for both LysoPLDs were basically the same.Conclusion:In this study, two different methods for improving soluble expression of human LysoPLD in E.coli were established and proved to be successful. The purified human LysoPLDs were enzymatically active, and the enzymatic activity were basically the same. -
磷脂酶是催化磷脂水解的第一步关键酶,根据水解磷脂的部位不同可以把磷脂酶分为磷脂酶A1、磷脂酶A2、磷脂酶 B、磷脂酶C和磷脂酶D[1]。磷脂酶D(PLD)是一个复杂的蛋白家族,普遍存在于哺乳动物、植物和细菌中[2-3]。磷脂酶D能够水解细胞膜中的磷脂,其水解产物参与细胞的多种生理过程如抗凋亡、伤口愈合、促进血管生成、神经系统发育、肿瘤进展等[4-5]。人溶血磷脂酶D(LysoPLD)因其与PLD的氨基酸序列具有相似性,也具有磷脂酶活性。作为PLD家族一员,它不仅可以水解溶血磷脂分子中磷酸二酯键生成溶血磷脂酸(LPA)和一个自由的头部基团,还能够催化磷脂和某些醇类发生碱基交换反应生成另一种磷脂[6-8]。
在食品工业中,PLD广泛作用于多种磷脂,以卵磷脂为底物,富集自然界稀少、难以分离提纯的单体磷脂,如磷脂酰丝氨酸(PS)、磷脂酰肌醇(PI)和磷脂酰甘油(PG)等,这些磷脂都具有非常高的经济价值[9-11]。如磷脂酰丝氨酸,是一种新资源食品,具有改善阿尔兹海默症、治疗抑郁症和缓解精神压力等作用,市场前景十分广阔[12-13]。酶法合成磷脂酰丝氨酸主要取决于PLD催化作用,但市售的PLD主要是从植物中提取,存在的问题主要是种类少、活力低、价格高。鉴于LysoPLD的PLD活性及其人源的低免疫原性,采用微生物异源表达LysoPLD具有解决市售PLD存在问题的潜力。
如何提升外源蛋白高效、可溶表达一直是工业酶生产与应用的瓶颈。目前,多个课题组尝试了磷脂酶D的异源表达,大多呈包涵体表达[14-15]。由于蛋白表达速度过快、活性二硫键来不及正确配对等原因,外源蛋白在大肠杆菌胞内往往会以无活性的包涵体形式表达。由于包涵体复性折叠机制不明确,把无活性的包涵体复性为完全的天然活性状态不仅操作繁琐而且复性效率较低。较低的复性效率依然是大肠杆菌体系表达的许多外源蛋白造成损失或者难以有效制备的主要原因[16]。原核表达系统及可溶性表达优化确实均属于非常成熟的技术,但在重组蛋白的表达过程中,合适的载体和分子伴侣对重组蛋白的正确折叠,防止包涵体的形成具有积极作用[17]。因此在实践过程中需要对不同标签蛋白和分子伴侣进行大量的筛选研究,确定合适的促溶表达条件。本研究首次尝试用触发因子(Triggering factor,Tig),作为第一个与新生多肽链相互作用的伴侣分子,以期协助LysoPLD蛋白共翻译折叠,有效促进其可溶表达。
前人通过定点突变研究发现,LysoPLD的活性中心(LysoPLD催化结构域)位于胞外结构域的Thr-210到His-475序列附近[18],确定含有Ser 48的C末端的胞外结构域是LysoPLD的主体,可以获得可溶、具有活性的重组分子[19]。在此基础上,本研究在大肠杆菌中分别构建含促溶标签MBP和分子伴侣tig共表达两种促LysoPLD可溶表达体系,并研究了两种促溶体系对重组蛋白的可溶表达、纯化方法、酶活性等方面的影响。
1. 材料与方法
1.1 材料与仪器
菌株和质粒大肠杆菌Escherichia coli DH5α、Escherichia coli BL21(DE3) 均由本实验室保存;pET28a-MBP质粒 由本实验保存;Chaperone Competent Cells pTf16/BL21 Takara公司;pET28a-MBP-PLD 本实验构建;限制性内切酶 美国NEB公司;胶回收试剂盒 美国Promega公司;质粒小提试剂盒 中国天根生物公司;SDS-PAGE电泳凝胶试剂盒、高保真PCR聚合酶 天津强微特生物科技有限公司;实验所用纯化填料 美国GE公司;麦芽糖等其他试剂 为国产分析纯。
AKTApurifier TM100纯化系统 美国GE公司;SCIE10TI-IID超声波细胞粉碎机 宁波新芝生物科技股份有限公司;Bio-radPowerPac HV垂直电泳仪 美国Bio-Rad公司;BioDoc-It2 315 Imaging System凝胶成像系统 美国UVP公司;GL-21M高速冷冻离心机 中国湘仪公司;UV3100紫外可见光分光光度计 日本岛津公司。
1.2 实验方法
1.2.1 人源LysoPLD基因的克隆及载体构建
根据NCBI上人LysoPLD(ENPP2)基因编码的氨基酸序列信息及催化反应的信息,确定本实验的目的克隆序列。通过信号肽分析程序(http://www.detaibio.com/tools /index.php?r=signal-peptide/index)对此序列进行信号肽分析,确定Ser-48至C末端全序列为本研究的目的基因序列。经密码子优化后,由通用生物合成目的基因序列,获得质粒pET28a-LysoPLD质粒。通过Snapgene设计麦芽糖结合蛋白(MBP)上下游引物,将MBP作为促溶标签与人LysoPLD基因融合表达,上游引物P1:5’-GTTTAACTTTAAGAAGGAGATATACC atgaaaatcgaagaaggtaaactg;下游引物P2:5’-AATATTGGTCCACGGACTCATATTGGATTGGAAGTACAGGTTTTCCTCGATCCCagtctgcgcgtctttcagggc;设计用于扩增pET28a-LysoPLD骨架的引物,上游引物P3:5’-ATGAGTCCGTGGACCAATATTAG;下游引物P4:5’-GGTATATCTCCTTCTTAAAGTTAAAC。
首先以P1、P2为引物对,PCR扩增MBP基因片段,再以P3、P4为引物对,PCR扩增pET28a-LysoPLD骨架,PCR的扩增条件为:98 ℃,3 min;(98 ℃,10 s;60 ℃,30 s;72 ℃,3.5 min) × 30循环;72 ℃,7 min;4 ℃,∞。将得到片段采用胶回收试剂盒进行片段回收,回收的片段以摩尔比1:3(MBP片段的摩尔数>pET28a-LysoPLD骨架的摩尔数)于室温采用同源重组的方法连接1 h,连接产物转化E.coli DH5α感受态,筛选阳性克隆,并进行测序鉴定。经鉴定的重组表达载体为pET28a-MBP-LysoPLD,转化E.coli BL21(DE3)感受态细胞后于−80 ℃进行保存[20]。
1.2.2 MBP-LysoPLD的诱导表达及条件优化
将−80 ℃保藏的重组表达菌株pET28a-MBP-PLD-BL21(DE3)复苏后,接种于5 mL含50 μg/mL卡那霉素的LB培养基中,37 ℃,100 r/min振荡过夜活化。将活化的pET28a-MBP-PLD-BL21(DE3)继续扩大培养,接种于100 mL含有卡那霉素的LB液体培养基中,当菌液A600达0.5~0.7时,加入IPTG诱导表达。6000 × g离心10 min收集菌体,超声破菌后,离心得到菌液上清和沉淀,再用0.5 mmol/L Tris-HCl将沉淀复溶,进行SDS-PAGE分析,配制10%分离胶和5%浓缩胶,恒定功率10 W跑45 min[21]。
1.2.3 Tig分子伴侣与PLD共表达实验
Takara公司的pTf16质粒中存在tig表达基因,受araB启动子调控,pET28a-LysoPLD质粒中目的基因LysoPLD受Lac调控系统控制,两个质粒具有不同的抗生素标记可进行共表达,且tig和LysoPLD可独立诱导表达[22]。将pET28a-LysoPLD和pTf16两个质粒同时转化BL21(DE3),采用双抗LB培养基(25 μg/mL卡那霉素和17 μg/mL氯霉素)筛选同时具有两种质粒的转化子,−80 ℃冻存。取出转化子,接种到双抗培养基中,37 ℃振荡培养活化;活化后,接种于100 mL含双抗和0.5 mg/mL L-Arabinose的LB培养基中进行扩大培养(此时已开始表达Tig),当A600达0.4~0.6时,于15 ℃放置30 min,加入终浓度为0.5 mmol/L IPTG于15 ℃振荡培养12~24 h。培养结束后,6000×g离心10 min收集菌体,超声破菌,根据SDS-PAGE电泳(具体方法同1.2.2)、活性测定结果确认目的产物的表达量及溶解性。
1.2.4 重组LysoPLD的纯化
MBP-LysoPLD的纯化:将诱导后收集的pET28a-MBP-LysoPLD的BL21(DE3)菌体均匀悬浮于50 mmol/L Tris-HCl(pH8.0)溶液中,经超声破菌后,20000×g离心20 min,收集上清,经0.45 μm滤膜过滤后,采用Amylose亲和柱进行纯化。采用Amylose-A(20 mmol/L Tris-HCl pH7.4,1 mmol/L EDTA,0.2 mol/L NaCl)进行洗涤、平衡,得到流穿蛋白,采用Amylose-B(20mmol/L Tris-HCl pH7.4,1 mmol/L EDTA,0.2 mol/LNaCl,10 mmol/L麦芽糖)进行洗脱,收集洗脱组分,得到洗脱收集蛋白,进行10%SDS-PAGE分析,具体方法同1.2.2。
LysoPLD(tig)的纯化:将诱导后收集的同时含有pTf16(tig)和pET28a-LysoPLD的BL21(DE3)菌体,均匀悬浮于50 mmol/L Tris-HCl(pH8.0)溶液中,经超声破碎后,20000×g离心20 min,收集上清,过0.45 μm滤膜。首先采用SP柱,经SP-A(20 mmol/L Tris-HCl(pH6.0))洗涤后,得到流穿蛋白。采用SP-B(20 mmol/L Tris-HCl(pH6.0),1 mol/L NaCl)进行洗脱,洗脱流速为1 mL/min,洗脱梯度为0~100% B,10 min收集洗脱组分,得到洗脱收集蛋白。再过Q柱纯化,经Q-A Buffer(20 mmol/L Tris-HCl pH8.0,1 mmol/L EDTA,5%Glycerol)洗涤后,得到流穿蛋白,采用Q-B Buffer(20 mmol/L Tris-HCl(pH8.0),1 mmol/L EDTA,5%Glycerol,1 mmol/L NaCl)进行洗脱,收集洗脱组分,得到洗脱收集蛋白,将上述得到的蛋白进行10%SDS-PAGE分析,具体方法同1.2.2。
1.2.5 LysoPLD的酶活测定
LysoPLD的酶活测定方法参考对硝基苯酚方法[23],对硝基苯酚粉末用50 mmol/L Tris-HCl (pH8.0)进行溶解并稀释,溶液浓度梯度为0、0.005、0.01、0.02和0.04 μmol/L,并测定其在波长为405 nm的分光光度值拟合得到对应标准曲线为y=28.36x,R2=0.9991。
取30 mg对硝基酚棕榈酸酯溶于10 mL异丙醇后,用含10% Triton-100的Tris-HCl(pH8.0)的溶液混合溶解,放入37 ℃恒温水浴5 min,加入适量酶液,测定405 nm处紫外分光光度值随时间变化的曲线,算出每分钟吸光值的值,酶水解活力计算公式为:
式中,△A为酶促反应的吸光变化值;V为测量试样总体积;Vs为所加酶液体积;K为对硝基酚标准曲线斜率;T为反应时间;277为转化系数。
1.2.6 最适温度、最适离子浓度和最适pH
将重组酶置于含有50 mmol/L CaCl2的40 mmol/L磷酸盐缓冲液(pH7.0)中,在20~80 °C温度范围内,每间隔10 °C进行酶反应,研究温度对重组酶活性的影响。
将50 μL20 mmol/L的重组酶添加到含有底物和100 μL 10、50和100 mmol/L不同金属离子(Zn2+、Ca2+和Mg2+)的溶液中后测量活性。另外,以40 mmol/L磷酸盐缓冲液(pH7.0)溶液为缓冲液进行对照。通过将不同因素的酶活性除以对照组的100%,计算出重组酶的相对活性(%)。
参照侯海娟等的方法,将纯化得到的两种磷脂酶D(10 μL)在50 °C下预孵育30 min,以含有对硝基酚棕榈酸酯的反应混合物为底物,以40 μL不同pH(5.0~8.5)缓冲液100 mmol/L乙酸盐、50 mmol/L三氯化甘油和8 mmol/L CaCl2作为缓冲液进行反应。
将20 mmol/L 50 μL的重组酶添加到含有底物对硝基酚棕榈酸酯和不同浓度(0、10、25、45和100 mmol/L)Ca2+离子(100 μL)的溶液中后测量活性。另外,以Triton X-100溶液为缓冲液进行对照。通过将不同因素的酶活性除以对照组的100%,计算出重组酶的相对活性(%)。
1.3 数据处理
实验中每个处理重复3次,采用SPSS 22.0软件进行数据显著性分析,应用Origin 8.0软件作图。
2. 结果与分析
2.1 pET28a-LysoPLD和pET28a-MBP-LysoPLD质粒的构建与鉴定
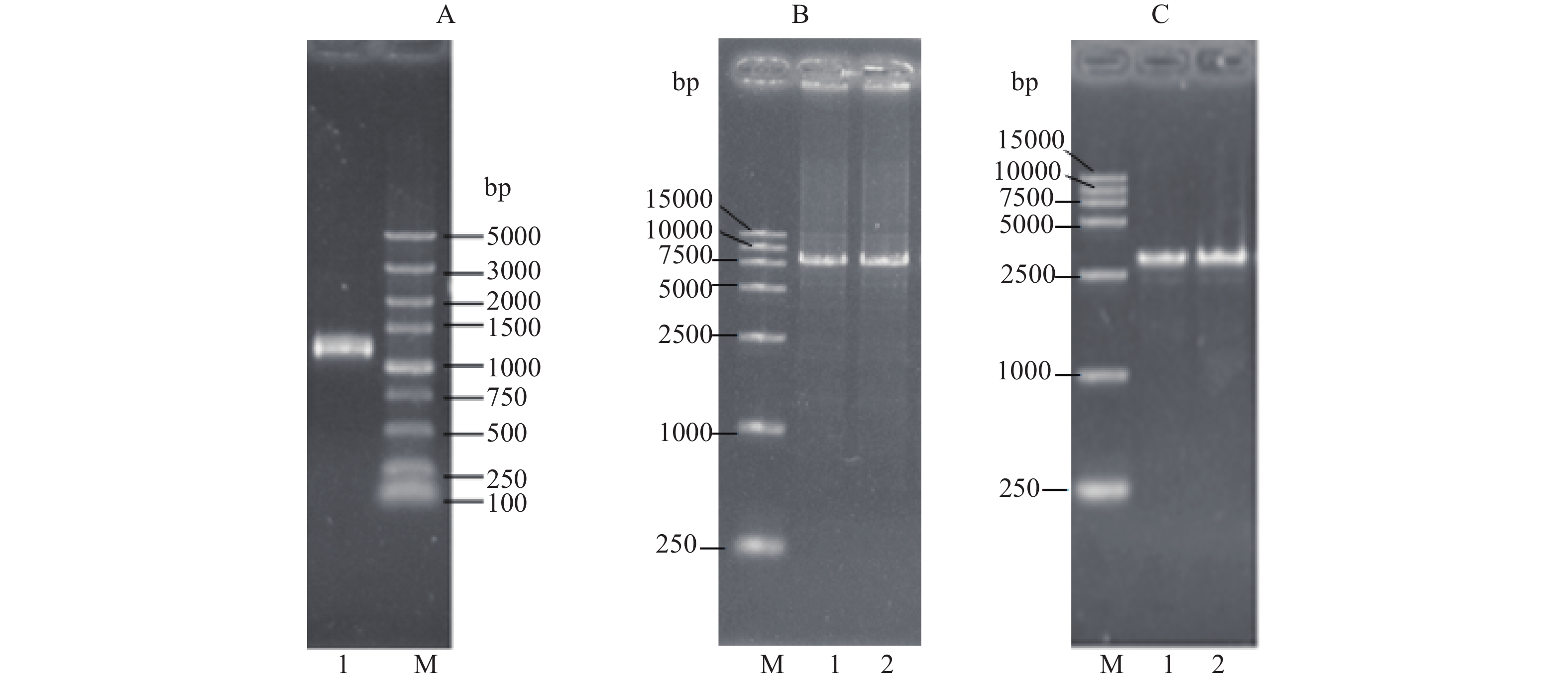
根据前述方法,通过PCR分别扩增MBP片段(~1100 bp,图1A)和pET28a-LysoPLD骨架(~7700 bp,图1B),同源重组后,获得的质粒采用T7/T7t引物对,对转化子进行PCR扩增鉴定,并采用1%琼脂糖凝胶电泳进行分析,出现~3500 bp的扩增产物,其大小恰好与MBP(~1100 bp) + LysoPLD(~2400 bp)大小相吻合(图1C)。重组质粒经抽提测序,BLAST比对,与目的基因序列相符,未发生突变,表明pET28a-MBP-LysoPLD载体构建成功。
![]() 图 1 MBP基因、pET28a-LysoPLD载体骨架和pET28a-MBP-LysoPLD基因的PCR扩增电泳图注:M:DNA Marker;泳道1(或2):PCR扩增的电泳条带;A:MBP基因的PCR扩增片段;B:pET28a-LysoPLD载体骨架的PCR扩增片段;C:pET28a-MBP-LysoPLD中PCR扩增片段鉴定电泳图。Figure 1. PCR amplification and identification of MBP gene、pET28a-LysoPLD and pET28a-MBP-LysoPLD
图 1 MBP基因、pET28a-LysoPLD载体骨架和pET28a-MBP-LysoPLD基因的PCR扩增电泳图注:M:DNA Marker;泳道1(或2):PCR扩增的电泳条带;A:MBP基因的PCR扩增片段;B:pET28a-LysoPLD载体骨架的PCR扩增片段;C:pET28a-MBP-LysoPLD中PCR扩增片段鉴定电泳图。Figure 1. PCR amplification and identification of MBP gene、pET28a-LysoPLD and pET28a-MBP-LysoPLD2.2 pET28a-MBP-LysoPLD-BL21(DE3)的诱导表达分析
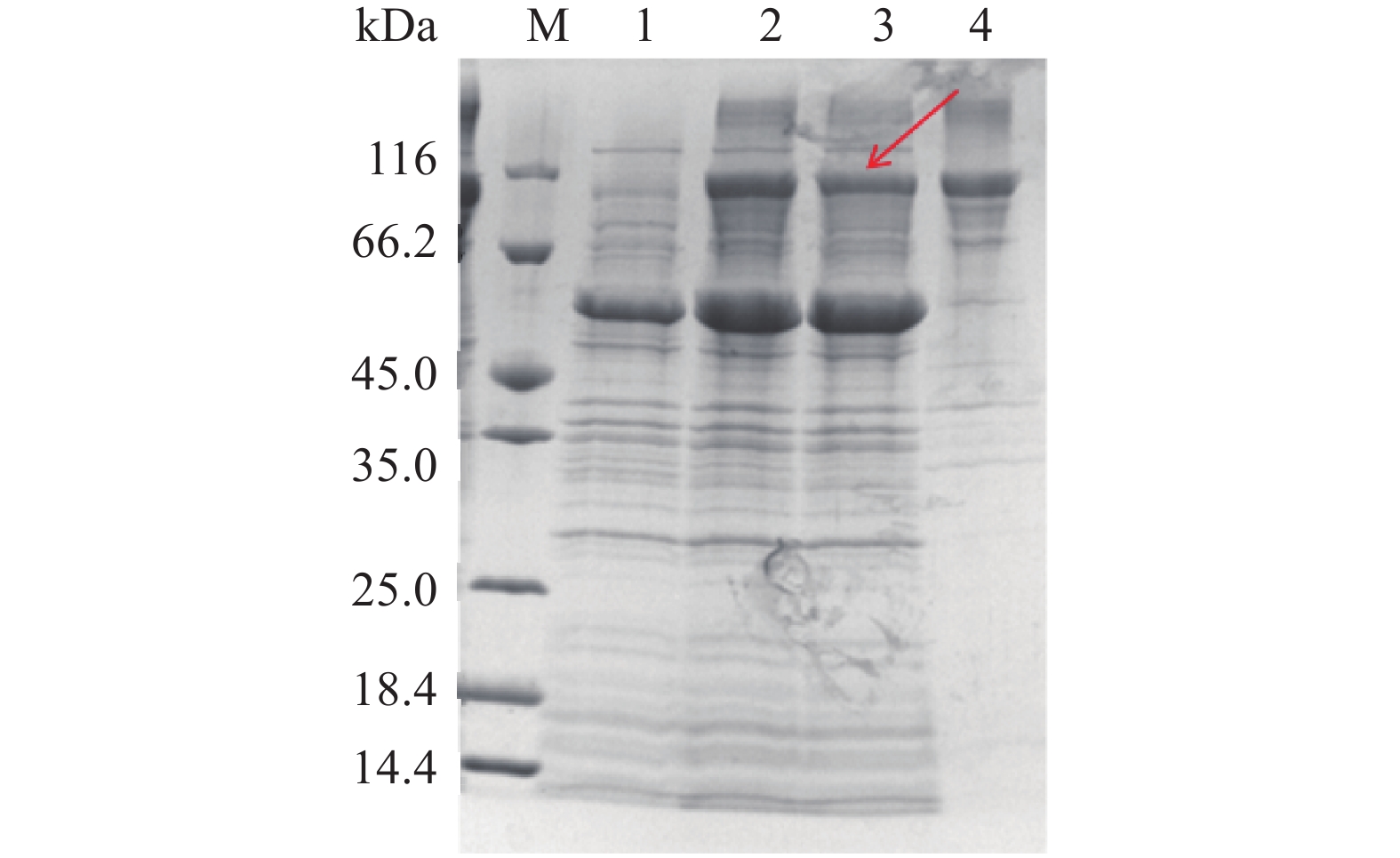
分别培养pET28a-LysoPLD-BL21(DE3)和pET28a-MBP-LysoPLD-BL21(DE3),并经IPTG诱导,收集菌体,超声破菌,进行SDS-PAGE电泳检测。如图2A可以看出,诱导后的全菌在110 kDa处有一明显蛋白质条带,但上清中却并未发现此蛋白的表达(图2A,泳道3),都在沉淀中(图2A,泳道4),证明没有MBP标签的LysoPLD蛋白几乎全部呈包涵体表达。MBP是一种大肠杆菌的内源性的蛋白质,由大肠杆菌K12的malE基因编码,分子量为40 kDa[24],在大肠杆菌中具有极高的表达量。在目的蛋白的氨基端添加MBP标签蛋白时,可大幅度地提高目的蛋白的可溶性表达量,并且可帮助目的蛋白在大肠杆菌中正确折叠并保持原有的活性[25]。因此。本实验构建了pET28a-MBP-LysoPLD,将LysoPLD蛋白与MBP进行融合表达,融合蛋白MBP-LysoPLD分子量大于116 kDa红色箭头标示的条带,由图2B显示,大约1/3的MBP-LysoPLD可在诱导表达后超声破碎离心得到的上清中表达(图2B,泳道4)。后续通过发酵工艺的优化,目的蛋白质的可溶表达量可能会有进一步的提高。
![]() 图 2 大肠杆菌表达的重组PLD和MBP-LysoPLD的SDS-PAGE分析注:A中泳道1~4分别代表大肠杆菌BL21(DE3)诱导表达前全菌,诱导表达后全菌,诱导表达后超声破碎离心得到的上清,诱导表达后超声破碎离心得到的沉淀;B中泳道1~4分别代表分别代表大肠杆菌BL21(DE3)诱导表达前全菌,诱导表达后全菌,诱导表达后超声破碎离心得到的沉淀,诱导表达后超声破碎离心得到的上清。Figure 2. Analysis of the recombinant of LysoPLD and MBP-LysoPLD expressed in E. coli by SDS-PAGE
图 2 大肠杆菌表达的重组PLD和MBP-LysoPLD的SDS-PAGE分析注:A中泳道1~4分别代表大肠杆菌BL21(DE3)诱导表达前全菌,诱导表达后全菌,诱导表达后超声破碎离心得到的上清,诱导表达后超声破碎离心得到的沉淀;B中泳道1~4分别代表分别代表大肠杆菌BL21(DE3)诱导表达前全菌,诱导表达后全菌,诱导表达后超声破碎离心得到的沉淀,诱导表达后超声破碎离心得到的上清。Figure 2. Analysis of the recombinant of LysoPLD and MBP-LysoPLD expressed in E. coli by SDS-PAGE2.3 Tig与LysoPLD的共诱导表达分析
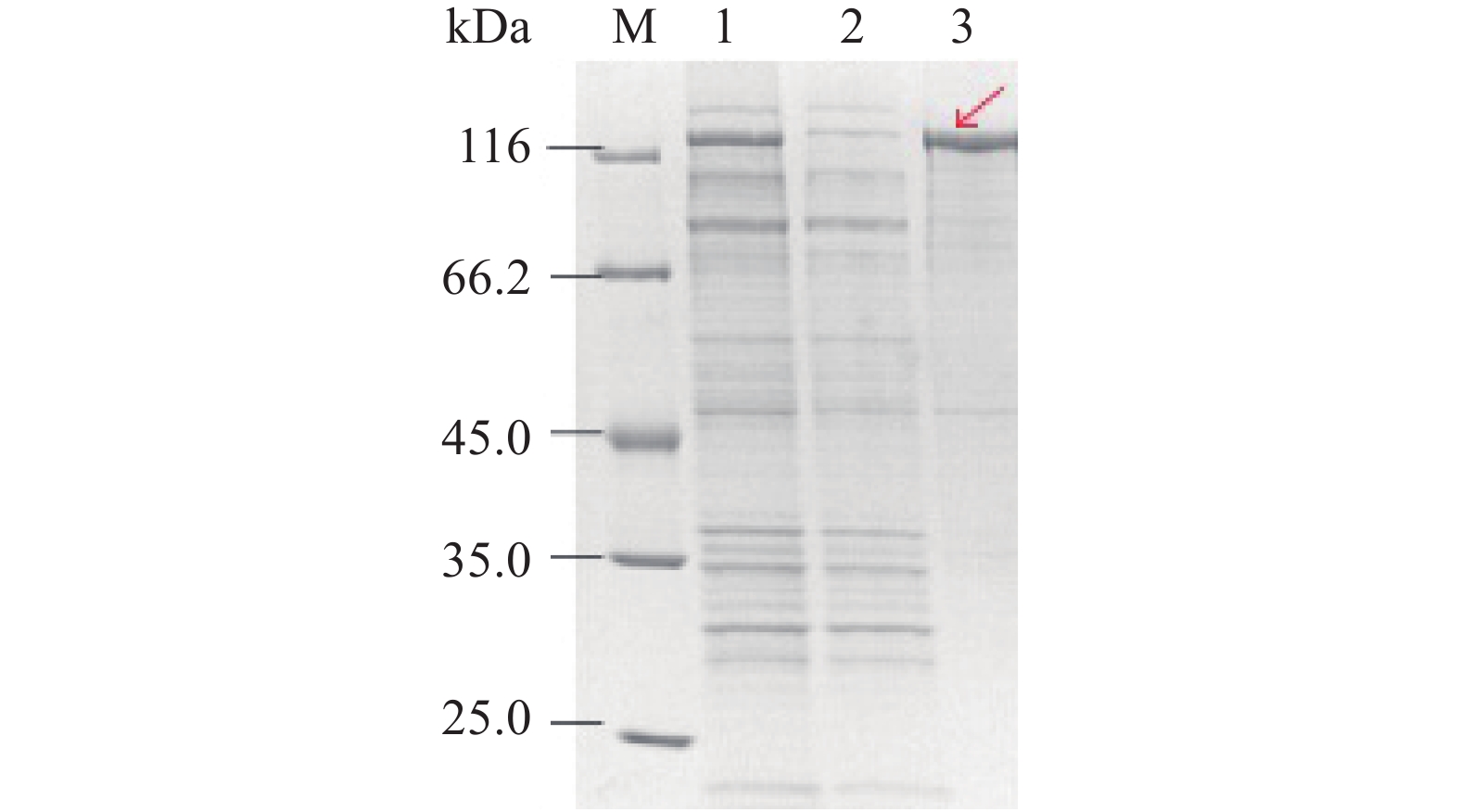
鉴于LysoPLD的原核表达主要以包涵体形式存在,除了用促溶标签的方法,本实验还尝试了共表达分子伴侣Tig的方法。如图3所示,在加入IPTG诱导前(图3,条带1)就已经在50 kDa左右出现了tig的诱导表达条带,在加入IPTG后,在66.2~116 kDa区间出现LysoPLD目的蛋白(图3,条带2),部分呈上清表达(图3,条带3),部分在破菌后离心沉淀中,即呈包涵体表达(图3,条带4),包涵体蛋白没有经过正确折叠的聚集体,不具备其应有的生物活性。对比SDS-PAGE图2B第4条泳道和图3中第3条泳道破菌离心的上清结果得到的可溶蛋白量大于促溶标签法。tig分子伴侣促溶法在诱导表达操作上较复杂,但经诱导表达后可直接获得LysoPLD蛋白本身。而促溶标签法诱导表达的是融合蛋白,后续需要把促溶标签通过蛋白酶进行切除后再纯化。
![]() 图 3 大肠杆菌表达的重组LysoPLD(tig)的SDS-PAGE分析注:M:Marker;1:诱导表达前全菌;2:诱导表达后全菌;3:诱导表达后超声破碎离心后得到的上清;4:诱导表达后超声破碎离心后得到的沉淀。Figure 3. Analysis of the recombinant of pTf16-LysoPLD(tig)expressed in E.coli by SDS-PAGE
图 3 大肠杆菌表达的重组LysoPLD(tig)的SDS-PAGE分析注:M:Marker;1:诱导表达前全菌;2:诱导表达后全菌;3:诱导表达后超声破碎离心后得到的上清;4:诱导表达后超声破碎离心后得到的沉淀。Figure 3. Analysis of the recombinant of pTf16-LysoPLD(tig)expressed in E.coli by SDS-PAGE2.4 LysoPLD(tig)和MBP-LysoPLD的分离纯化比较分析
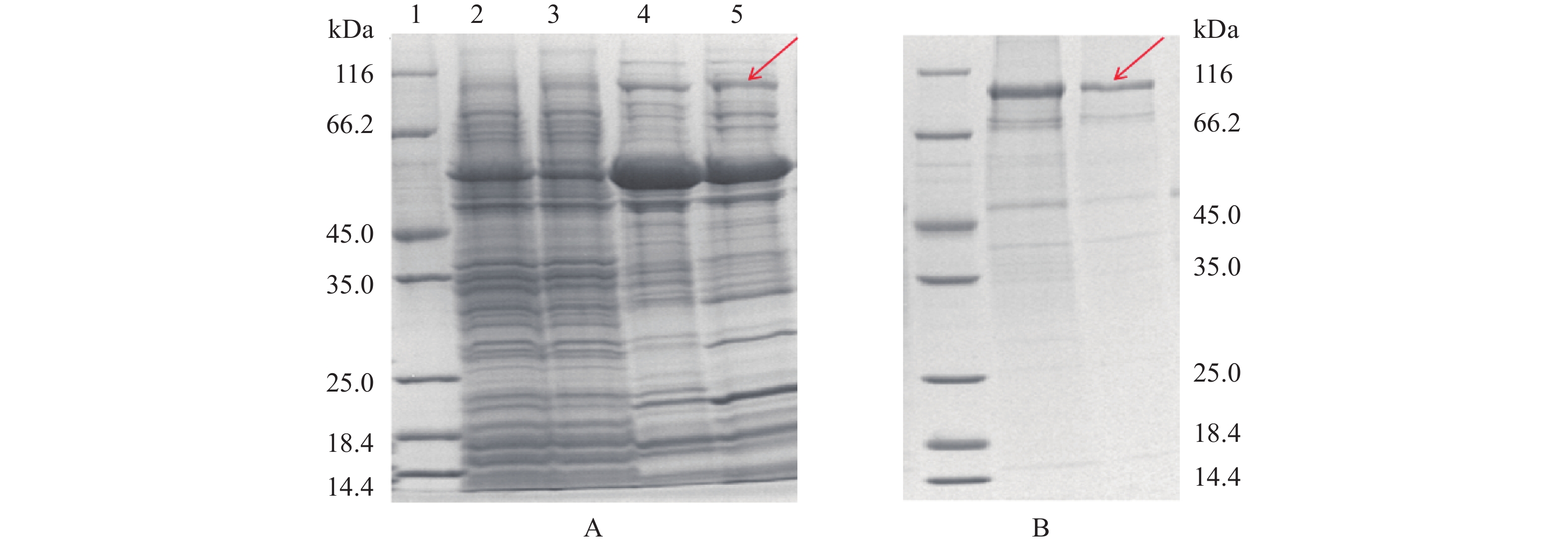
根据MBP标签特性,本实验采用了Amylose亲和柱层析对MBP-LysoPLD融合蛋白的纯化,目标蛋白条带大于116 kDa,对Amylose亲和柱吸附特异性强,可以被洗脱,结果如图4箭头所示,最终条带3为目标蛋白MBP-LysoPLD,所得最终样品约4 mL,浓度0.46 mg/mL,根据凝胶成像分析系统分析其纯度达到90%以上。
![]() 图 4 Amylose Resin纯化洗脱MBP-LysoPLD注:M:Marker;1:上Amylose亲和柱前总蛋白;2:经Amylose亲和柱流穿的蛋白;3:经Amylose-B洗脱收集的蛋白。Figure 4. Purification and elution of MBP-LysoPLD by Amylose Resin
图 4 Amylose Resin纯化洗脱MBP-LysoPLD注:M:Marker;1:上Amylose亲和柱前总蛋白;2:经Amylose亲和柱流穿的蛋白;3:经Amylose-B洗脱收集的蛋白。Figure 4. Purification and elution of MBP-LysoPLD by Amylose Resintig协助下的LysoPLD蛋白表达菌株经超声破碎,细胞上清液依次经过SP琼脂糖凝胶柱(图5A)和阴离子Q柱(图5B),进行目的蛋白洗脱,最终获得分子量为110 kDa的目标蛋白(图5B)。在设计LysoPLD的表达时,PLD序列设计了His标签,但无论His标签在C端或在N端,PLD蛋白都无法挂Ni柱(结果未示出),因此采用离子交换柱Q柱进行PLD蛋白的纯化,根据凝胶成像分析系统分析其纯度达到80%以上。
![]() 图 5 蛋白纯化后的SDS-PAGE电泳图注:A:SP琼脂糖凝胶柱LysoPLD蛋白洗脱SDS-PAGE电泳图;泳道1~5分别为1:Marker;2:上SP琼脂糖凝胶柱前总蛋白;3:经SP琼脂糖凝胶柱流穿的蛋白;4:经SP-B洗脱后收集的蛋白;5:经SP-B洗脱后收集的蛋白;(B):阴离子柱(Q柱)LysoPLD蛋白洗脱SDS-PAGE图;1,Marker;2,经Q-B洗脱后收集的蛋白;3,Q-B洗脱后收集的蛋白。Figure 5. SDS-PAGE analysis of purified PLD protein
图 5 蛋白纯化后的SDS-PAGE电泳图注:A:SP琼脂糖凝胶柱LysoPLD蛋白洗脱SDS-PAGE电泳图;泳道1~5分别为1:Marker;2:上SP琼脂糖凝胶柱前总蛋白;3:经SP琼脂糖凝胶柱流穿的蛋白;4:经SP-B洗脱后收集的蛋白;5:经SP-B洗脱后收集的蛋白;(B):阴离子柱(Q柱)LysoPLD蛋白洗脱SDS-PAGE图;1,Marker;2,经Q-B洗脱后收集的蛋白;3,Q-B洗脱后收集的蛋白。Figure 5. SDS-PAGE analysis of purified PLD protein比较tig协助下的LysoPLD蛋白和融合蛋白MBP-LysoPLD的分离纯化步骤发现,MBP-LysoPLD由于存在MBP标签,只采用Amylose亲和层析就可以得到纯度在90%的融合蛋白,但后续也会涉及采用TEV进行MBP标签切割步骤;而tig协助下的LysoPLD蛋白由于不能挂Ni柱纯化,需要进行两步离子柱洗脱,在操作方法上来讲较为复杂,但不需要再进一步切除多余的标签。因此,从纯化步骤的角度两种方法复杂度类似。
2.5 LysoPLD(tig)与MBP-LysoPLD的酶活比较分析
采用同样100 mL发酵液体积,MBP-LysoPLD融合蛋白得到4.91 mg,tig协助折叠的LysoPLD蛋白得到5.79 mg,tig协助的PLD蛋白比融合蛋白MBP-PLD得率约高17.9%。根据1.2.5中的酶活测定方法,分别测定融合蛋白MBP-LysoPLD和tig协助折叠的PLD的酶活力,MBP-LysoPLD的总活力为20.98 U,比活力为4.27 U/mg,LysoPLD(tig)总活力为42.61 U,比活力为7.32 U/mg(表1)。
表 1 两种LysoPLD酶活性质比较Table 1. Comparison of two LysoPLD enzyme activity指标 MBP-PLD LysoPLD(tig) 总蛋白量(mg) 4.91 ± 0.03 5.79 ± 0.11 总活力(U) 20.98 ± 2.34 42.61 ± 3.05 比酶活(U/mg) 4.27 ± 0.02 7.32 ± 1.24 目前报道的LysoPLD蛋白的表达系统有Sf 9昆虫细胞和COS-7细胞表达[19],现有结果表明这两类表达系统表达的蛋白缺乏修饰,蛋白表达量低,无法满足LysoPLD蛋白结构及功能研究,也有研究者对LysoPLD蛋白进行HEK293细胞的规模化表达,但其表达的LysoPLD蛋白含有FC标签,标签较大,后续需要切去标签,获得蛋白纯度低[26]。本试验构建的LysoPLD(tig)可以有效避免切标签带来的操作,与其他来源的标签相比,表达效率高,可溶性效果好,具有重要的应用价值。
2.6 pH、温度及金属离子对LysoPLD酶活的影响比较
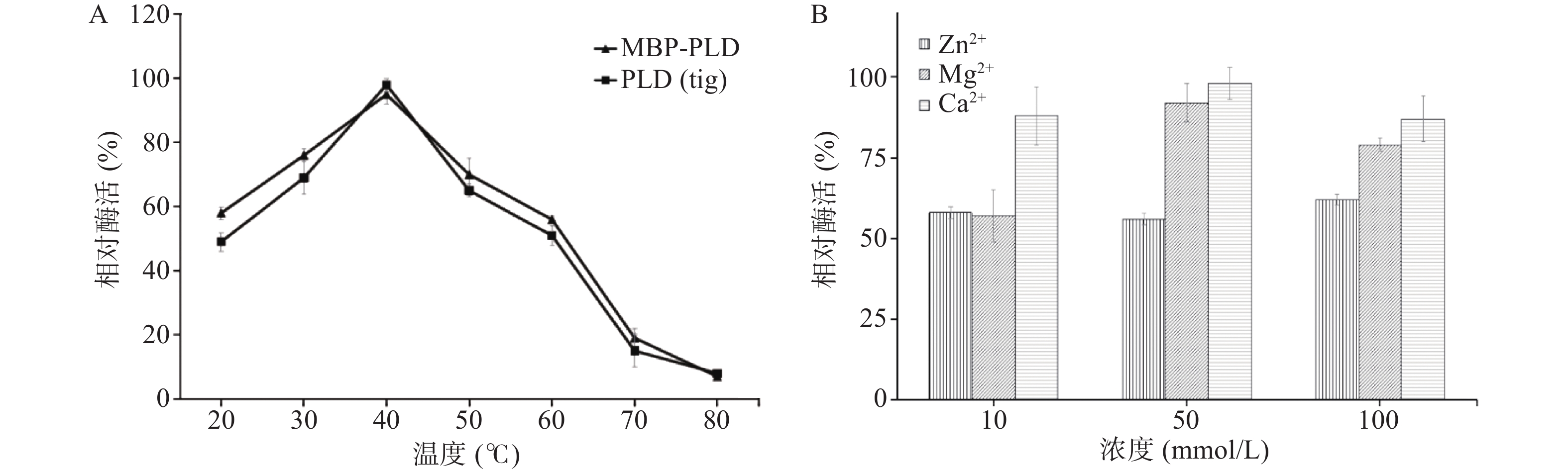
图6(A)表明LysoPLD在pH为5.5,钙离子浓度为50 mmol/L时,其相对酶活的最适温度为40 ℃。温度达到60 ℃仍保留50%的酶活力,高于60 ℃时活性降低并逐渐消失。故LysoPLD的温度耐受范围在20~60 ℃。不同浓度的金属离子处理LysoPLD蛋白,检测其对LysoPLD活性影响(图6B)可知,不同金属离子明显影响重组酶的活性,且呈现浓度依赖性,尤其是50 mmol/L钙离子时,重组酶活性最高,与重组酶有Ca2+的结合位点有关,与相关文献的研究结果一致[27]。
![]() 图 6 LysoPLD(tig)与MBP-LysoPLD在不同温度相对酶活(A)和LysoPLD(tig)在不同金属离子下相对酶活(B)Figure 6. Relative enzyme activities of LysoPLD (tig) and MBP-LysoPLD at different temperature (A) and LysoPLD (tig) at different metal ions (B)
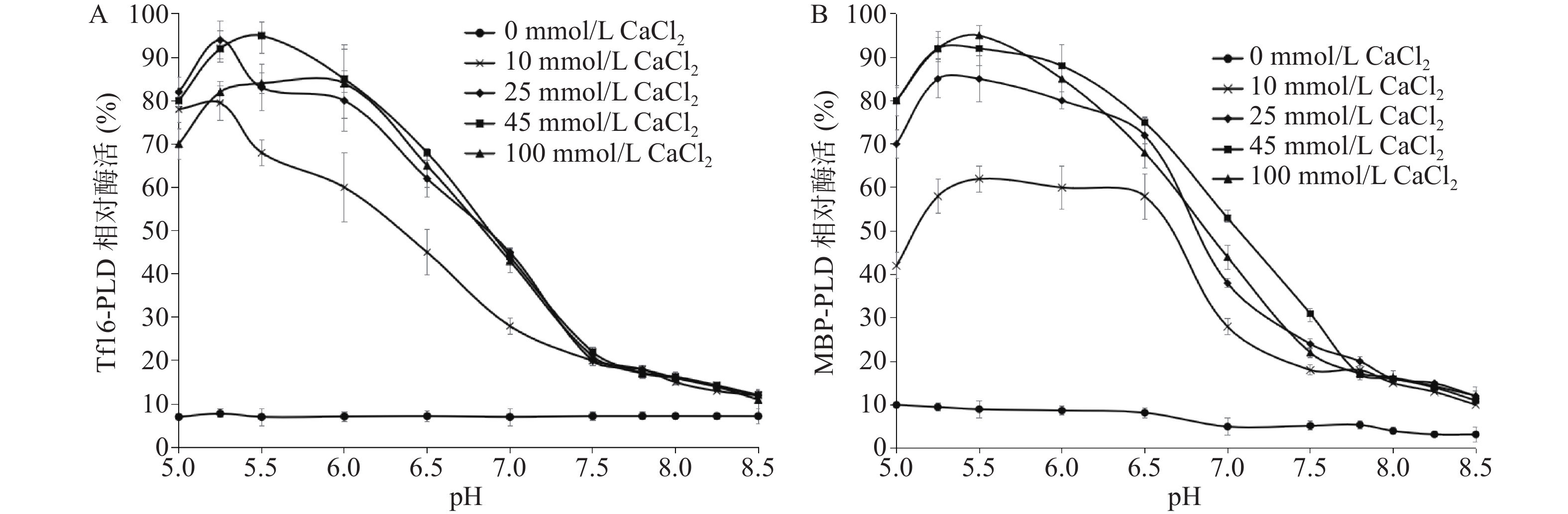
图 6 LysoPLD(tig)与MBP-LysoPLD在不同温度相对酶活(A)和LysoPLD(tig)在不同金属离子下相对酶活(B)Figure 6. Relative enzyme activities of LysoPLD (tig) and MBP-LysoPLD at different temperature (A) and LysoPLD (tig) at different metal ions (B)LysoPLD的酶催化功能依赖于Ca2+离子,同时pH对其酶活的影响也很大,因此,本研究对比了LysoPLD(tig)和MBP-LysoPLD在Ca2+离子依赖性及最佳pH方面的特性[28](图7)。结果发现,两种LysoPLD均呈现显著Ca2+的依赖性。随着pH的提高,在不同钙离子浓度作用下,两种PLD的相对酶活均呈现先增高后降低,且LysoPLD(tig)和MBP-LysoPLD二者的最佳pH都在5.5~5.6之间,最佳酶活时需要的钙离子浓度是45 mmol/L。因此,通过PLD(tig)及MBP-LysoPLD蛋白的酶活特性比较分析可以发现,在最适pH和最适钙离子浓度条件下,两种酶的相对酶活均达到90%以上,因此,促溶标签法和tig共表达方法都可获得发挥正常功能,具有一致特性的LysoPLD。
![]() 图 7 PLD(tig)与MBP-LysoPLD在不同pH和浓度Ca2+下相对酶活Figure 7. Relative enzyme activities of LysoPLD (tig) and MBP-LysoPLD at different pH and Ca2+ concentration
图 7 PLD(tig)与MBP-LysoPLD在不同pH和浓度Ca2+下相对酶活Figure 7. Relative enzyme activities of LysoPLD (tig) and MBP-LysoPLD at different pH and Ca2+ concentration3. 结论
成功构建了人PLD的原核表达质粒pET28a-pTf16-LysoPLD(tig)和pET28a-MBP-LysoPLD,建立了LysoPLD(tig)和MBP-LysoPLD蛋白大肠杆菌促进可溶性表达系统,获得了纯度80%以上的LysoPLD蛋白。两种LysoPLD酶反应最适pH在5.5~5.6之间,最佳酶活时需要的钙离子浓度是45 mmol/L,两种促进可溶性表达方式都可以获得具有一致特性的LysoPLD。
-
![]()
图 1 MBP基因、pET28a-LysoPLD载体骨架和pET28a-MBP-LysoPLD基因的PCR扩增电泳图
注:M:DNA Marker;泳道1(或2):PCR扩增的电泳条带;A:MBP基因的PCR扩增片段;B:pET28a-LysoPLD载体骨架的PCR扩增片段;C:pET28a-MBP-LysoPLD中PCR扩增片段鉴定电泳图。
Figure 1. PCR amplification and identification of MBP gene、pET28a-LysoPLD and pET28a-MBP-LysoPLD
![]()
图 2 大肠杆菌表达的重组PLD和MBP-LysoPLD的SDS-PAGE分析
注:A中泳道1~4分别代表大肠杆菌BL21(DE3)诱导表达前全菌,诱导表达后全菌,诱导表达后超声破碎离心得到的上清,诱导表达后超声破碎离心得到的沉淀;B中泳道1~4分别代表分别代表大肠杆菌BL21(DE3)诱导表达前全菌,诱导表达后全菌,诱导表达后超声破碎离心得到的沉淀,诱导表达后超声破碎离心得到的上清。
Figure 2. Analysis of the recombinant of LysoPLD and MBP-LysoPLD expressed in E. coli by SDS-PAGE
![]()
图 3 大肠杆菌表达的重组LysoPLD(tig)的SDS-PAGE分析
注:M:Marker;1:诱导表达前全菌;2:诱导表达后全菌;3:诱导表达后超声破碎离心后得到的上清;4:诱导表达后超声破碎离心后得到的沉淀。
Figure 3. Analysis of the recombinant of pTf16-LysoPLD(tig)expressed in E.coli by SDS-PAGE
![]()
图 4 Amylose Resin纯化洗脱MBP-LysoPLD
注:M:Marker;1:上Amylose亲和柱前总蛋白;2:经Amylose亲和柱流穿的蛋白;3:经Amylose-B洗脱收集的蛋白。
Figure 4. Purification and elution of MBP-LysoPLD by Amylose Resin
![]()
图 5 蛋白纯化后的SDS-PAGE电泳图
注:A:SP琼脂糖凝胶柱LysoPLD蛋白洗脱SDS-PAGE电泳图;泳道1~5分别为1:Marker;2:上SP琼脂糖凝胶柱前总蛋白;3:经SP琼脂糖凝胶柱流穿的蛋白;4:经SP-B洗脱后收集的蛋白;5:经SP-B洗脱后收集的蛋白;(B):阴离子柱(Q柱)LysoPLD蛋白洗脱SDS-PAGE图;1,Marker;2,经Q-B洗脱后收集的蛋白;3,Q-B洗脱后收集的蛋白。
Figure 5. SDS-PAGE analysis of purified PLD protein
![]()
图 6 LysoPLD(tig)与MBP-LysoPLD在不同温度相对酶活(A)和LysoPLD(tig)在不同金属离子下相对酶活(B)
Figure 6. Relative enzyme activities of LysoPLD (tig) and MBP-LysoPLD at different temperature (A) and LysoPLD (tig) at different metal ions (B)
![]()
图 7 PLD(tig)与MBP-LysoPLD在不同pH和浓度Ca2+下相对酶活
Figure 7. Relative enzyme activities of LysoPLD (tig) and MBP-LysoPLD at different pH and Ca2+ concentration
表 1 两种LysoPLD酶活性质比较
Table 1 Comparison of two LysoPLD enzyme activity
指标 MBP-PLD LysoPLD(tig) 总蛋白量(mg) 4.91 ± 0.03 5.79 ± 0.11 总活力(U) 20.98 ± 2.34 42.61 ± 3.05 比酶活(U/mg) 4.27 ± 0.02 7.32 ± 1.24  下载: 导出CSV
下载: 导出CSV
-
[1] 叶展, 罗质, 何东平, 等. 酶法脱胶及其在大豆油适度精炼中的应用[J]. 食品工业,2015,36(1):258−261. [2] 刘茂希. 缺氧微环境中HIF1-α上调PLD2表达进而抑制结肠癌细胞凋亡的机制研究[D]. 重庆: 重庆医科大学, 2016. [3] 陈雷. 玉米磷脂酶D基因家族的克隆及功能分析[D]. 雅安: 四川农业大学, 2017. [4] Hong Y Y, Zhao J, Guo L, et al. Plant phospholipases D and C and their diverse functions in stress responses[J]. Progress in Lipid Research,2016,62:55−74. doi: 10.1016/j.plipres.2016.01.002
[5] Cholia R P, Nayyar H, Kumar R, et al. Understanding the multifaceted role of ectonucleotide pyrophosphatase/ phosphodiesterase 2 (ENPP2) and its altered behaviour in human diseases[J]. Current Molecular Medicine,2015,15(10):932−943. doi: 10.2174/1566524015666150921104804
[6] Koike S, Keino-Masu K, Ohto T, et al. The N-terminal hydrophobic sequence of autotaxin(ENPP2) functions as a signal peptide[J]. Genes to Cells: Devoted to Molecular & Cellular Mechanisms,2006,11(2):133−142.
[7] Magotti P, Bauer I, Igarashi M, et al. Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: Regulation of fatty acid ethanolamide biosynthesis by bile acids[J]. Structure (London, England: 1993),2015,23(3):598−604. doi: 10.1016/j.str.2014.12.018
[8] Ueda N, Minami K, Ishimoto K, et al. Effects of lysophosphatidic acid (LPA) receptor-2 (LPA2) and LPA3 on the regulation of chemoresistance to anticancer drug in lung cancer cells[J]. Cellular Signalling,2020,69:109551. doi: 10.1016/j.cellsig.2020.109551
[9] 陈晓玲. 黑曲霉磷脂酶D基因的克隆表达与生化特征解析[D]. 天津: 天津科技大学, 2018. [10] Hou H J, Gong J S, Dong Y X, et al. Phospholipase D engineering for improving the biocatalytic synthesis of phosphatidylserine[J]. Bioprocess and Biosystems Engineering,2019,42(7):1185−1194. doi: 10.1007/s00449-019-02116-7
[11] Zhou W B, Gong J S, Hou H J, et al. Mining of a phospholipase D and its application in enzymatic preparation of phosphatidylserine[J]. Bioengineered,2018,9(1):80−89. doi: 10.1080/21655979.2017.1308992
[12] 李冰麟. 磷脂酶D催化磷脂酰基转移反应生产稀有及非天然磷脂的研究[D]. 西安: 西北大学, 2019. [13] 侯海娟, 龚劲松, 翟珅, 等. 磷脂酶D的重组表达及其在磷脂酰丝氨酸合成中的应用[J]. 食品与发酵工业,2019,45(13):9−14. [14] Yang L Y, Xu Y, Chen Y, et al. Efficient extracellular expression of phospholipase D in Escherichia coli with an optimized signal peptide[J]. IOP Conference Series: Materials Science and Engineering,2018,301:012105. doi: 10.1088/1757-899X/301/1/012105
[15] Ciervo A, Mancini F, Cassone A. Transcription, expression, localization and immunoreactivity of Chlamydophila pneumoniae phospholipase D protein[J]. Microbial Pathogenesis,2007,43(2/3):96−105.
[16] 何伟. 重组大肠杆菌表达包涵体药物蛋白的复性与纯化研究[D]. 上海: 华东理工大学, 2017. [17] 李祥魁, 范翠英, 崔亚君, 等. 重组蛋白可溶性表达促进标签的研究进展[J]. 生物技术,2013,23(2):93−97. doi: 10.3969/j.issn.1004-311X.2013.02.049 [18] Gijsbers R, Aoki J, Arai H, et al. The hydrolysis of lysophospholipids and nucleotides by autotaxin (NPP2) involves a single catalytic site[J]. FEBS Letters,2003,538(1/2/3):60−64.
[19] Haga A, Hashimoto K, Tanaka N, et al. Scalable purification and characterization of the extracellular domain of human autotaxin from prokaryotic cells[J]. Protein Expression and Purification,2008,59(1):9−17. doi: 10.1016/j.pep.2007.12.008
[20] Hannes M B, Patrick G, Sophia L. et al. AQUA cloning: A versatile and simple enzyme-free cloning approach[J]. Plos One,2015,10(9):e0137652. doi: 10.1371/journal.pone.0137652
[21] Piao D C, Shin D W, Kim I S, et al. Trigger factor assisted soluble expression of recombinant spike protein of porcine epidemic diarrhea virus in Escherichia coli[J]. BMC Biotechnology,2016,16(1):39. doi: 10.1186/s12896-016-0268-7
[22] Nishihara K, Kanemori M, Yanagi H, et al. Overexpression of trigger factor prevents aggregation of recombinant proteins in Escherichia coli[J]. Applied and Environmental Microbiology,2000,66(3):884−889. doi: 10.1128/AEM.66.3.884-889.2000
[23] 张莹. 在不同宿主中表达链霉菌磷脂酶D的研究[D]. 广州: 华东理工大学, 2013. [24] Lebendiker M, Danieli T. Purification of proteins fused to maltose-binding protein[M]//Methods in Molecular Biology. New York, NY: Springer New York, 2016: 257−273.
[25] 张亚萌. 重组融合蛋白rMBP-NAP的毒理学研究[D]. 郑州: 郑州大学, 2016. [26] Song Y D, Dilger E, Bell J, et al. Large scale purification and characterization of recombinant human autotaxin/ lysophospholipase D from mammalian cells[J]. BMB Reports,2010,43(8):541−546. doi: 10.5483/BMBRep.2010.43.8.541
[27] 赵亚蕊, 曹鹏程, 郑耀武, 等. 人自分泌运动因子哺乳细胞表达纯化及酶学特性[J]. 生物技术,2017,27(5):484−490. [28] Yang H Y, Roberts M F. Cloning, overexpression, and characterization of a bacterial Ca2+-dependent phospholipase D[J]. Protein Science,2009,11(12):2958−2968. doi: 10.1110/ps.0225302

 下载:
下载:








计量
- 文章访问数: 285
- HTML全文浏览量: 121
- PDF下载量: 21
